26. Mai 2023
Philips legt neue Testergebnisse der CPAP/BiPAP-Schlaftherapiegeräte vor
Amsterdam, Niederlande – Royal Philips (NYSE: PHG, AEX: PHIA) informiert heute über den aktuellen Stand des umfassenden Test- und Forschungsprogramms seiner Tochtergesellschaft Philips Respironics zur Bewertung potenzieller Gesundheitsrisiken im Zusammenhang mit dem schalldämpfenden Schaumstoff auf Polyesterbasis (PE-PUR) in bestimmten Schlaftherapie- und Beatmungsgeräten, die unter die freiwillige Rückrufmeldung/Sicherheitsmitteilung vom Juni 2021 fallen. [1] Jetzt wurden die Risikobewertungen für die CPAP/BiPAP-Schlaftherapiegeräte abgeschlossen, die unter die Rückrufmeldung/Sicherheitsmitteilung [1] fallen. Dazu gehören die DreamStation, System One und die DreamStation Go der ersten Generation. Sie machen einen Anteil von etwa 95 Prozent der weltweit registrierten Geräte aus. Die Bewertungen bauen auf den vorangegangenen Aktualisierungen auf – veröffentlicht im Dezember 2021, Juni 2022 und Dezember 2022. Zusätzlich wurden Analysen und Tests für DreamStation-Geräte der ersten Generation durchgeführt, die einer Ozonreinigung unterzogen wurden.
Testmethoden
Das Test- und Forschungsprogramm wurde in Zusammenarbeit mit fünf unabhängigen, zertifizierten Testlaboren durchgeführt. Qualifizierte, unabhängige Expertinnen und Experten, Philips Respironics sowie ein externes medizinisches Gremium haben die Ergebnisse anschliessend geprüft und bewertet. Die angewandten Testmethoden – von der Testplanung über die Durchführung bis hin zur Interpretation der Ergebnisse für die durchgeführten Risikobewertungen – entsprechen den geltenden Industrienormen ISO 18562 [2] und ISO 10993 [3]. Das Design der angewandten Testmethoden wurde zudem wissenschaftlich untermauert, basierend auf einer gründlichen Berücksichtigung und Abmilderung von Testbeschränkungen, die jeder Testnorm und/oder wissenschaftlichen Forschung innewohnen. So wurden beispielsweise Tests an mehreren gebrauchten Geräten mit unterschiedlichem Patientengebrauch und sichtbarer visueller Schaumstoffdegradation sowie an im Labor gealtertem Schaumstoff durchgeführt, der absichtlich in unterschiedlichem Masse degradiert worden war. Bei den Risikobewertungen wurden sehr konservative Annahmen zugrunde gelegt. Weitere Beispiele finden sich am Ende dieser Pressemitteilung. „Unsere oberste Priorität sind die Gesundheit und das Wohlbefinden der Patientinnen und Patienten “, so Roy Jakobs, CEO von Royal Philips. „Wir haben uns daher auf dieses umfassende Test- und Forschungsprogramm konzentriert, um mehr Klarheit über die Sicherheit der betroffenen Geräte zu gewinnen, sowie auf die Bereitstellung von Ersatzgeräten für die Patientinnen und Patienten. Die heute vorgestellten Risikobewertungen für die Schlaftherapiegeräte sind positiv und beruhigend und wir machen gute Fortschritte bei der Remedierung der betroffenen Geräte. Die zuständigen Behörden auf der ganzen Welt, einschliesslich der FDA, prüfen die Testergebnisse und Bewertungen noch. Wir verfolgen das gleiche Ziel, nämlich die Sicherheit und Qualität der Gesundheitsversorgung für Patientinnen und Patienten zu gewährleisten, und wir werden weiterhin eng mit diesen Behörden zusammenarbeiten. Der Abschluss der Tests und die Remedierung der betroffenen Geräte haben für uns weiterhin höchste Priorität.“
Testergebnisse und Analysen für Schlaftherapiegeräte, die nicht mit Ozon gereinigt wurden
Die abgeschlossenen Testergebnisse und Analysen für die Schlaftherapiegeräte der ersten Generation DreamStation, System One und DreamStation Go deuten darauf hin, dass eine potenzielle Exposition der Patientinnen und Patienten mit Schaumstoffpartikeln (PM) und flüchtigen organischen Verbindungen (VOCs) aus dem PE-PUR-Schaumstoff in diesen Geräten wahrscheinlich nicht zu einer nennenswerten Gesundheitsbeeinträchtigung führt. Die durchgeführten Tests und Schlussfolgerungen sind in der nachstehenden Tabelle zusammengefasst.
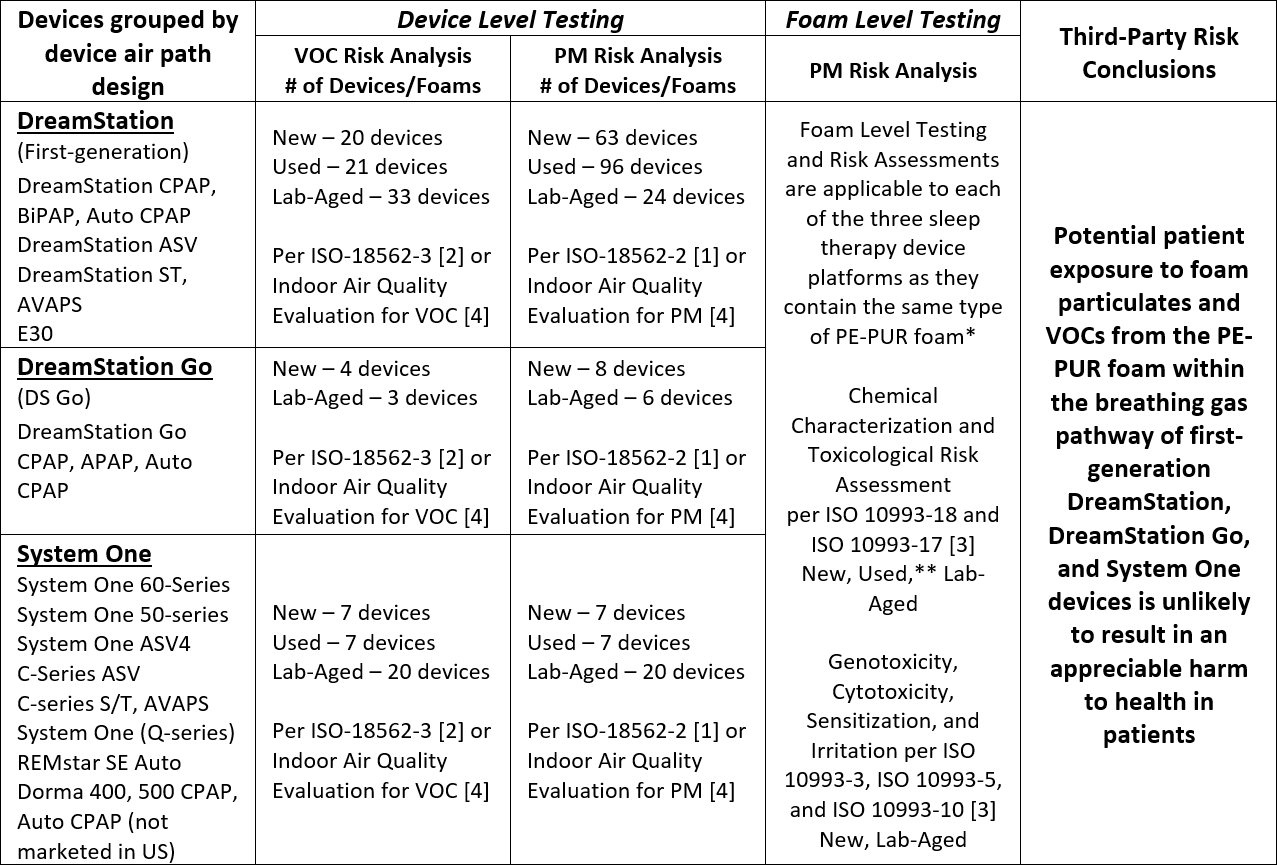
VOC: Flüchtige organische Verbindungen PM: Schaumstoffpartikel * Die Gesamtmenge des Schaums in den Geräten variiert je nach Auslegung und Konfiguration des Luftweges zwischen etwa 1 g und 10 g. Die Geräte innerhalb der einzelnen Plattformen haben die gleiche Luftführung und Konfiguration, einschliesslich der vorhandenen Schaummenge. ** Der Schaum von 7 verschiedenen gebrauchten DreamStation-Geräten der ersten Generation wurde gemäss ISO 10993-18 und -173 chemisch charakterisiert und enthielt Schaum, der für eine Reihe von visuellen Degradationszuständen repräsentativ ist.
Darüber hinaus unterschieden sich die getesteten Schaumstoffpartikel-Emissionen von gebrauchten Geräten mit sichtbarer Degradation statistisch nicht von denen gebrauchten Geräten ohne Degradation, was darauf hindeutet, dass die Degradation nicht zu einer nennenswerten Erhöhung lungengängiger Partikel in den getesteten Geräten beigetragen hat.
Selbst unter der sehr konservativen und theoretischen Annahme, dass sich der gesamte Schaumstoff zersetzen könnte und dass man dann dem gesamten zersetzten PE-PUR-Schaumstoff in den Geräten ausgesetzt ist, kam die Risikobewertung durch unabhängige Dritte zu dem Schluss, dass die Exposition gegenüber Partikeln aus zersetztem Schaumstoff in diesen Geräten, einschliesslich potenziell lungengängiger und nicht lungengängiger Partikel, wahrscheinlich nicht zu einer nennenswerten gesundheitlichen Beeinträchtigung der Patientinnen und Patienten führen wird.
Die Auswirkungen der Ozonreinigung auf den Schaumstoffabbau bei DreamStation-Geräten der ersten Generation
Philips Respironics hat Tests mit DreamStation-Geräten der ersten Generation abgeschlossen, die einer Ozonreinigung unterzogen wurden.
Zusammenfassung der laufenden Tests
Philips Respironics ist dabei, verschiedene verbleibende Tests und Analysen abzuschliessen. Die Risikobewertungen für System One- und DreamStation Go-Geräte (die den gleichen Schaumstoff wie die DreamStation-Geräte der ersten Generation enthalten), die mit Ozon gereinigt wurden, werden derzeit abgeschlossen. Für die Beatmungsgeräte Trilogy 100/200 und OmniLab Advanced Plus werden die VOC- und Schaumstoffpartikel-Tests sowie die chemische Bewertung und toxikologische Risikobewertung fortgesetzt. Diese Geräte enthalten eine andere Art von PE-PUR-Schaum als die DreamStation-Geräte der ersten Generation. [5] Philips Respironics wird voraussichtlich im dritten Quartal 2023 ein Update zu diesem Thema vorlegen.
Hinweise für Leistungserbringende und Patientinnen und Patienten
Patientinnen und Patienten, die derzeit ein betroffenes Schlaftherapiegerät verwenden, das noch nicht remediert wurde und noch nicht registriert ist, werden gebeten, ihre Geräte zu registrieren, um die Remedierung zu erleichtern. Philips Respironics rät Patientinnen und Patienten, die ein betroffenes Gerät verwenden, das noch nicht remediert wurde, sich mit ihrer Ärztin oder ihrem Arzt oder Leistungserbringende in Verbindung zu setzen, um über eine geeignete Vorgehensweise zu entscheiden. Diese kann darin bestehen, die Verwendung des Geräts einzustellen, das betroffene Gerät weiter zu verwenden, ein anderes ähnliches Gerät zu verwenden, das nicht Teil der Sicherheitsmitteilung ist, oder alternative Behandlungen für Schlafapnoe anzuwenden. Darüber hinaus wird den Patientinnen und Patienten geraten, die Anweisungen und empfohlenen Reinigungs- und Austauschrichtlinien von Philips Respironics für ihr Schlaftherapiegerät und das Zubehör zu befolgen. Ozon- und UV-Licht-Reinigungsprodukte sind derzeit keine zugelassenen Reinigungsmethoden für Schlaftherapiegeräte oder -masken und sollten nicht verwendet werden. Philips Respironics rät Benutzerinnen und Benutzern von Beatmungsgeräten auch weiterhin, sich mit ihren medizinischen Ansprechpersonen in Verbindung zu setzen, bevor sie ihre Therapie ändern.
Wissenschaftliche Untermauerung der Testmethoden
Die Entwicklung der angewandten Prüfmethoden wurde wissenschaftlich untermauert, indem die Prüfbeschränkungen, die jeder Prüfnorm und/oder wissenschaftlichen Forschung innewohnen, sorgfältig berücksichtigt und angepasst wurden. Zur Veranschaulichung werden im Folgenden Beispiele für derartige Überlegungen aufgeführt.
Bei der wissenschaftlichen Untermauerung der angewandten Prüfmethoden wurden beispielsweise folgende Prüfbeschränkungen erwogen:
¹ Freiwillige Rückrufmeldung in den USA / Sicherheitsmitteilung für den Rest der Welt ² ISO 18562-2: Bewertung der Biokompatibilität der Atemgaswege bei medizinischen Anwendungen – Teil 2: Prüfung für Emissionen von Partikeln; ISO 18562-3: Bewertung der Biokompatibilität der Atemgaswege bei medizinischen Anwendungen – Teil 3: Prüfungen für Emissionen von flüchtigen organischen Verbindungen ³ ISO 10993: Biologische Bewertung von Medizinprodukten: Teil 1: Bewertung und Prüfung im Rahmen eines Risikomanagementprozesses, Teil 3: Prüfungen auf Genotoxizität, Karzinogenität und Reproduktionstoxizität, Teil 5: Prüfungen auf In-vitro-Zytotoxizität, Teil 10: Prüfungen auf Irritationen und Sensibilisierung der Haut, Teil 17: Nachweis zulässiger Grenzwerte für herauslösbare Bestandteile, Teil 18: Chemische Charakterisierung von Werkstoffen für Medizinprodukte im Rahmen eines Risikomanagementprozesses ⁴ Die Norm, die für Prüfungen vor der ISO 18562 verwendet wurde. ⁵ DreamStation-, SystemOne- und DreamStation Go-Geräte der ersten Generation enthalten PE-PUR-Schaumstoff des Typs A, während Trilogy 100/200-Geräte PE-PUR-Schaumstoff des Typs B und OmniLab Advanced Plus-Geräte PE-PUR-Schaumstoffe der Typen A und B enthalten. Die bekannten Unterschiede zwischen den Schaumstoffen des Typs A und des Typs B bestehen darin, dass der Schaumstoff des Typs B mit einem druckempfindlichen Acrylklebstoff verwendet werden kann, eine geringere Dichte und eine andere Dicke aufweist und ausserdem einen Zusatzstoff enthält, der die potenzielle Entflammbarkeit verringert.
Über Royal Philips
Royal Philips (NYSE: PHG, AEX: PHIA) ist ein führender Anbieter im Bereich der Gesundheitstechnologie. Ziel des Unternehmens mit Hauptsitz in den Niederlanden ist es, die Gesundheit und das Wohlbefinden der Menschen zu verbessern und sie mit entsprechenden Produkten und Lösungen in allen Phasen des Health Continuums zu begleiten: während des gesunden Lebens, aber auch in der Prävention, Diagnostik, Therapie sowie der häuslichen Pflege. Die Entwicklungsgrundlagen dieser integrierten Lösungen sind fortschrittliche Technologien sowie ein tiefgreifendes Verständnis für die Bedürfnisse von medizinischem Fachpersonal, Konsumentinnen und Konsumenten. Das Unternehmen ist führend in diagnostischer Bildgebung, bildgestützter Therapie, Patientenmonitoring und Gesundheits-IT sowie bei Gesundheitsprodukten für Verbraucherinnen und Verbraucher und in der häuslichen Pflege. Philips beschäftigt etwa 74.000 Mitarbeiterinnen und Mitarbeiter in mehr als 100 Ländern und erzielte 2022 einen Umsatz von 17,8 Milliarden Euro. Mehr über Philips im Internet: www.philips.ch/healthcare

")
")